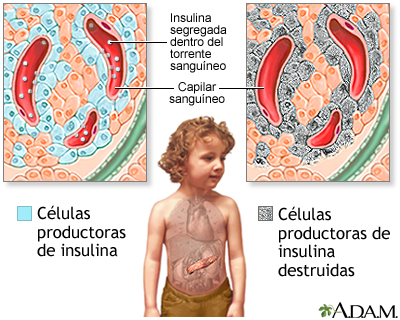
Recordemos que, ante la falta de insulina, la glucosa va a entrar a las neuronas, porque la entrada de glucosa a las neuronas no necesita insulina (a diferencia de su entrada a a otros tejidos como el hígado y el músculos, que si esta mediada por insulina). Toda la glucosa que necesita receptor para entra a otros tejidos no podrá entrar y, entonces se va a las neuronas, donde se convierte en sorbitol. El sorbitol va a causar daño osmótico a nivel periférico en la células de Shwam. El daño osmótico produce desmielinizacion, y esto se manifiesta como NEUROPATIAS DIABETICAS. Ante esta condición, el paciente va a presentar reducción/perdida de la sensibilidad en forma de calcetines y guantes, a lo que denominamos "acropaestesia".
Ademas de lo mencionado, el paciente va a presentar gastroparesia (perdida de sensibilidad en el estomago), que se va a manifestar como disminución de la motilidad y retraso del vaciamiento gástrico, razón por la cual el paciente se va a sentir satisfecho por mucho tiempo.
También, se presenta enteropatía diabética, el paciente tiene problemas/trastornos con la motilidad del intestino, por lo que va a presentar dos cosas: estreñimiento o diarrea.
Fuente: ¡Mis apuntes de clase!